
[GROMACS] adalah package berkecepatan tinggi untuk komputasi dinamika molekular baik untuk biokimia (seperti protein, lipids) maupun non-biokimia (seperti polimer).
Modul GROMACS
Berikut adalah detail versi GROMACS yang tersedia secara global di ALELEON Supercomputer:
Modul GROMACS versi CPU
| Versi
|
Nama modul software
|
Dukungan hardware
|

|

|
OMP
Threading
|
MPI
multi-node
|
| 2021.1
|
gromacs/2021.1-GCC10-MKL
|
V
|
-
|
-
|
V
|
Modul GROMACS versi CPU dan GPU
| Versi
|
Nama modul software
|
Dukungan hardware
|
|
|
|
OMP
Threading
|
MPI
multi-node
|
| 2022.4
|
gromacs/2022.4-GCC11.3-AOCL4.0-CUDA11.8
|
V
|
V
|
V
|
V
|
| 2024.4
|
gromacs/2024.4-GCC12.3-AOCL5.0-CUDA12.5
|
V
|
V
|
V
|
V
|
Hubungi tim admin melalui email support@efisonlt.com untuk permintaan instalasi versi lain.
Info Menjalankan GROMACS dengan Akselerasi GPU
ALELEON Supercomputer menyediakan GROMACS yang dapat berjalan dengan akselerasi GPU. Beberapa hal perlu menjadi pertimbangan user:
- Akselerasi GPU bukan jaminan peningkatan performa pada semua jenis input dan parameter.
- Tim admin menyarankan user untuk menguji hal ini di perangkat komputer pribadi.
- Biasanya GROMACS mdrun tidak memerlukan parameter tambahan untuk akselerasi GPU.
- Dalam beberapa kasus, optimasi parameter tambahan dapat meningkatkan kecepatan komputasi.
- Referensi lanjutan parameter akselerasi GPU (-nb, -bonded, -pme) dapat dipelajari pada tautan berikut:
Pilihan Menjalankan Komputasi
User dapat menjalankan GROMACS dengan pilihan metode:
- [Batch Job] -> menjalankan komputasi melalui manajemen Slurm di [compute node].
(klik expand / kembangkan)
Batch Job
Tersedia dua pilihan tampilan yaitu terminal dan web EFIRO melalui app Job Composer. Pilih sesuai keinginan.
| - Langkah Batch Job di Terminal -
|
| 0
|
Menggunakan terminal!
- User web EFIRO buka app
Aleleon Shell Access.
|
| 1
|
| Lakukan persiapan sebelum batch job:
|
- Siapkan file komputasi yang dibutuhkan.
- Pre-processing input dan parameter GROMACS lihat subbab:
- [GROMACS - Pre Processing Input dan Parameter]
|
| 2A
|
Buat Submit Script yaitu 'formulir' untuk menjalankan job komputasi.
- Nama file bebas dengan format .sh, contoh
submit.sh
|
|
|
Contoh template Submit Script
- Ikuti petunjuk NOTES dan alur script di dalamnya.
Klik expand / kembangkan
| - Template Submit Script GROMACS versi CPU -

|
#!/bin/bash
# -----------------------------------------------------
# Template SLURM Submit Script
# GROMACS (versi CPU) | rev.201224
#
# NOTES:
# 1. Isi bagian yang ditandai 4 garing (////).
# 2. Template ini bersifat referensi.
# User dapat mengubah bagian yang perlu diubah.
# -----------------------------------------------------
# -----------------------------------------------------
# Alokasi komputasi SBATCH dan file input
# -----------------------------------------------------
# Alokasi jumlah core thread CPU
#SBATCH --ntasks=////
# Alokasi jumlah memori RAM (satuan GB)
#SBATCH --mem=////GB
# Alokasi limit waktu menjalankan job
# Format HH:MM:SS atau D-HH:MM:SS
#SBATCH --time=////
# Definisi file untuk menampung output terminal program
#SBATCH --output=result-%j.txt
# Definisi file untuk menampung output error log
#SBATCH --error=error-%j.txt
# Parameter input dan output untuk gmx_mpi mdrun
GROMACS_PARAMETER="////"
# ----------------------------------------------------
# Script jalannya program
# ----------------------------------------------------
# Memuat modul software GROMACS
# Daftar modul lihat subbab "Modul GROMACS" di atas
module load gromacs////
# Perintah menjakankan GROMACS
mpirun -np ${SLURM_NTASKS} gmx_mpi mdrun ${GROMACS_PARAMETER}
|
| - Template Submit Script GROMACS versi GPU -

|
 Note: Note:
- GROMACS versi GPU menggunakan threading OMP per task MPI:
- Task MPI diwakili oleh jumlah GPU (
SBATCH gpus)
- Thread OMP diwakili oleh
SBATCH cpus-per-task
- Perhatikan total CPU = ntasks * cpus-per-task
- Jumlah thread OMP disarankan 2 atau 4.
|
#!/bin/bash
# --------------------------------------------------------
# Template SLURM Submit Script
# GROMACS (versi GPU) | rev.201224
#
# NOTES:
# 1. Isi bagian yang ditandai 4 garing (////).
# 2. Template ini bersifat referensi.
# User dapat mengubah bagian yang perlu diubah.
# 3. GROMACS GPU menggunakan threading OMP per task MPI
# Perhatikan usage CPU = ntasks * cpus-per-task
# --------------------------------------------------------
# -----------------------------------------------------
# Alokasi komputasi SBATCH dan file input
# -----------------------------------------------------
# Menggunakan partisi compute node GPU
#SBATCH --partition=ampere
# Alokasi jumlah core thread CPU
#SBATCH --ntasks=////
# Alokasi jumlah GPU, menjadi jumlah task MPI
#SBATCH --gpus=////
# Alokasi jumlah thread OMP per task MPI
# Disarankan 2 atau 4
#SBATCH --cpus-per-task=////
# Alokasi jumlah memori RAM (satuan GB)
#SBATCH --mem=////GB
# Alokasi limit waktu menjalankan job
# Format HH:MM:SS atau D-HH:MM:SS
#SBATCH --time=////
# Definisi file untuk menampung output terminal program
#SBATCH --output=result-%j.txt
# Definisi file untuk menampung output error log
#SBATCH --error=error-%j.txt
# Parameter input dan output untuk gmx_mpi mdrun
GROMACS_PARAMETER="////"
# ----------------------------------------------------
# Script jalannya program
# ----------------------------------------------------
# Memuat modul software GROMACS yang dapat menggunakan GPU
# Daftar modul lihat subbab "Modul GROMACS" di atas
module load gromacs////
# Threading OMP
export OMP_NUM_THREADS=${SLURM_CPUS_PER_TASK}
# Perintah menjalankan GROMACS 1 GPU dan multiple GPU
if [ $SLURM_GPUS -eq 1 ]
then
echo "Terdeteksi running GROMACS dengan 1 GPU"
gmx_mpi mdrun ${GROMACS_PARAMETER}
elif [ $SLURM_GPUS -gt 1 ]
then
echo "Terdeteksi running GROMACS dengan $SLURM_GPUS GPU"
# Unset UCX supaya MPI berjalan di NVIDIA
unset UCX_TLS
unset UCX_NET_DEVICES
mpirun -np $SLURM_GPUS gmx_mpi mdrun ${GROMACS_PARAMETER}
fi
|
|
- Untuk SBATCH notifikasi email status jalannya job lihat Slurm Jojo.
|
| 2B
|
SBATCH komputasi (ntasks, mem, time, dll) punya limit berdasarkan:
- Sisa Core Hour user dan [Fair Usage Limit]
- Spesifikasi sistem dan software komputasi.
Untuk melihat limit, jalankan perintah berikut:
$ slimit-gromacs
Limitasi perintah:
- Jalankan ketika tidak ada job user yang sedang berjalan atau antri.
|
| 3
|
Jalankan job komputasi dengan perintah:
$ sbatch [nama-submit-script]
Contoh:
$ sbatch submit.sh
|
| 4
|
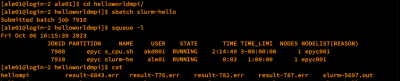
User dapat melihat status jalannya job dengan perintah:
$ squeue -ul $USER
Kolom ST atau STATE menunjukkan status jalannya job:
| STATE
|
Penjelasan
|
R (RUN)
|
Job berjalan
|
PD (PENDING)
|
Job tertahan, lihat NODELIST(REASON)
|
CG (COMPLETING)
|
Job selesai dan dalam proses clean-up
|
CA (CANCELED)
|
Job dibatalkan user
|
PR (PREEMPETED)
|
Job dibatalkan admin, alasan dikabarkan via email
|
S (SUSPENDED)
|
Job ditahan admin, alasan dikabarkan via email
|
|
| 5
|
| User dapat memantau file output dengan perintah Unix seperti:
|
| 6
|
Apabila user ingin menghentikan job yang berjalan, jalankan perintah:
$ scancel [job-ID]
Job ID ada pada squeue diatas.
contoh membatalkan job ID 231:
$ scancel 231
|
| ---
|
| - Langkah Batch Job di EFIRO Job Composer -
|
| 1
|
| Login ke web EFIRO ALELEON Supercomputer.
|
| 2
|
 Buka pinned apps Job Composer di homepage EFIRO. Buka pinned apps Job Composer di homepage EFIRO.
- Atau melalui menu
Apps > Job Composer
|
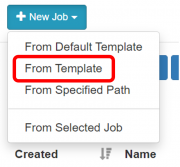
Pilih menu New Job > From Template
|
| 3
|
| Buat ruang job untuk menjalankan komputasi:
|
Pillih template GROMACS yang diinginkan:
| Nama Template
|
Penjelasan
|
| GROMACS 2021.1 (CPU)
|
Menjalankan GROMACS 2021.1 pada CPU
|
| GROMACS 2022.4 (CPU)
|
Menjalankan GROMACS 2022.4 pada CPU
|
| GROMACS 2022.4 (GPU)
|
Menjalankan GROMACS 2022.4 pada GPU
|
| GROMACS 2024.4 (CPU)
|
Menjalankan GROMACS 2022.4 pada CPU
|
| GROMACS 2024.4 (GPU)
|
Menjalankan GROMACS 2022.4 pada GPU
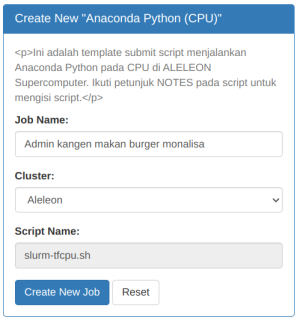
| Kemudian isi Job Name dan klik Create New Job
|
| 4
|
| Lakukan persiapan sebelum batch job:
|
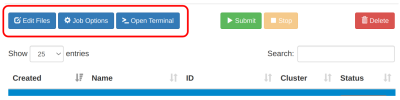
Siapkan file komputasi yang dibutuhkan di ruang job.
- Pastikan ruang job yang dipilih adalah yang akan digunakan.
- Upload / download / edit file via menu
Edit Files
- Upload / download file tunggal diatas 2GB gunakan [software FTP]
Pre-processing input dan parameter via menu Open Terminal
- Lihat subbab [GROMACS - Pre Processing Input dan Parameter]
|
| 5A
|
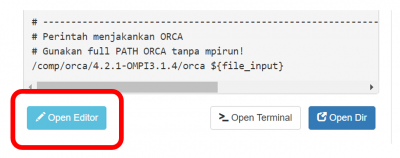
Lengkapi Submit Script melalui tombol Open Editor
- Submit script adalah 'formulir' untuk menjalankan job komputasi.
- Ikuti petunjuk NOTES didalamnya.
- Klik
Save setiap kali mengubah script.
|
Bagi yang memilih template GROMACS versi GPU:
- GROMACS versi GPU menggunakan threading OMP per task MPI:
- Task MPI diwakili oleh jumlah GPU (
SBATCH gpus)
- Thread OMP diwakili oleh
SBATCH cpus-per-task
- Perhatikan total CPU = ntasks * cpus-per-task
- Jumlah thread OMP disarankan 2 atau 4.
|
|
|
|
|
| 5B
|
SBATCH komputasi (ntasks, mem, time, dll) punya limit berdasarkan:
- Sisa Core Hour user dan Fair Usage Limit.
- Spesifikasi sistem dan software komputasi.
Untuk melihat limit, jalankan perintah berikut via Open Terminal
$ slimit-gromacs
Limitasi perintah:
- Jalankan ketika tidak ada job user yang sedang berjalan atau antri.
|
| 6
|
Jalankan job dengan klik tombol Submit
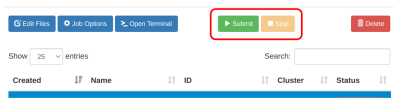
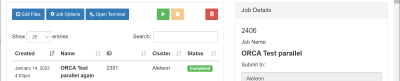
Pantau kolom Status yang menjelaskan status jalannya job:
| Status
|
Arti
|
| Not Submitted
|
Job belum pernah dijalankan.
|
| Running
|
Job berjalan.
|
| Queue
|
Job mengantri dan belum berjalan.
|
| Completed
|
Job selesai berjalan.
|
| Failed
|
Job berhenti karena error atau di stop user.
|
| Untuk melihat alasan job yang queue:
|
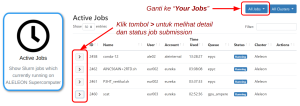 Buka pinned apps Active Jobs di homepage EFIRO. Ubah opsi All jobs ke Your Jobs. Klik simbol > untuk melihat status job yang pending pada kolom Reason. Arti reason lihat laman Daftar Reason NODELIST ALELEON Supercomputer. |
|
| 7
|
Lihat output file komputasi pada kolom Folder Contents
- Atau dengan membuka menu
Edit Files
|
| 8
|
Apabila user ingin menghentikan job yang berjalan, klik Stop
|
| 9
|
Untuk menghapus ruang job dan datanya:
- Pilih ruang job yang akan dihapus.
- Klik menu
Delete
|
| ---
|
Pre-processing Input dan Parameter
GROMACS membutuhkan pengolahan file input dan parameter sebelum menjalankan komputasi.
- Kegiatan ini dilakukan di login node sehingga tidak mengkonsumsi Kredit Core Hour.
- Hubungi tim admin apabila user membutuhkan panduan atau tambahan software package tertentu.
ALELEON Supercomputer menyediakan variasi berikut:
- [gmx_mpi]
- [ACPYPE]
gmx_mpi
GROMACS ALELEON Supercomputer diinstal dengan dukungan MPI sehingga user menggunakan gmx_mpi, bukan gmx.
| Menjalankan gmx_mpi
|
| 0
|
Menggunakan terminal!
- User web EFIRO buka app
Aleleon Shell Access.
- Atau menu
Open Terminal pada ruang job Composer.
|
| 1
|
Aktifkan modul GROMACS yang akan dipakai.
$ ml [nama-modul-GROMACS]
Nama modul lihat subbab 'Modul GROMACS' diatas.
|
| 2
|
Gunakan perintah gmx_mpi untuk pre-processing input.
Contoh dengan pdb2gmx:
$ gmx_mpi pdb2gmx [parameter-input-output]
|
| ---
|
Kembali ke subbab [Batch Job]
ACPYPE (AnteChamber PYthon Parser InterfacE)
ACPYPE adalah wrapper Python berdasarkan software ANTECHAMBER untuk membantu pengolahan file input dan parameter software MD (Molecular Dynamic) seperti GROMACS ini.
| - Menggunakan container ACPYPE -
|
| 0
|
Menggunakan terminal!
- User web EFIRO buka app
Aleleon Shell Access.
- Atau menu
Open Terminal pada ruang job Composer.
|
| 1
|
Aktifkan container Apptainer:
$ ml apptainer
|
| 2
|
Aktifkan container ACPYPE, tersedia versi 2023.10.27:
$ apptainer run $SCONT/acpype-2023.10.27.sif
Kemudian user dapat menggunakan ACPYPE:
Apptainer> [jalankan perintah ACPYPE ...]
|
| 3
|
Untuk keluar dari container, jalankan perintah:
Apptainer> exit
|
| ---
|
Kembali ke subbab [Batch Job]
Pelaporan Kendala dan Support
Apabila menjumpai masalah teknis dalam menjalankan komputasi, silahkan lapor dengan klik gambar berikut ini.

Bila terdapat pertanyaan lainnya, silahkan hubungi admin EFISON melalui email:
support@efisonlt.com






 Buka pinned apps Job Composer di homepage EFIRO.
Buka pinned apps Job Composer di homepage EFIRO.








