
GROMACS adalah package berkecepatan tinggi untuk komputasi dinamika molekular baik untuk biokimia (seperti protein, lipids) maupun non-biokimia (seperti polimer).
Modul GROMACS
Berikut adalah detail versi GROMACS yang tersedia secara global di ALELEON Supercomputer:
Modul GROMACS versi CPU
| Versi
|
Nama modul software
|
Dukungan hardware
|
| CPU
|
GPU
|
OMP
Threading
|
MPI
multi-node
|
| 2021.1
|
gromacs/2021.1-GCC10-MKL
|
V
|
-
|
-
|
V
|
Modul GROMACS versi CPU dan GPU
| Versi
|
Nama modul software
|
Dukungan hardware
|
| CPU
|
GPU
|
OMP
Threading
|
MPI
multi-node
|
| 2022.4
|
gromacs/2022.4-GCC11.3-AOCL4.0-CUDA11.8
|
V
|
V
|
V
|
V
|
| 2024.4
|
gromacs/2024.4-GCC12.3-AOCL5.0-CUDA12.5
|
V
|
V
|
V
|
V
|
Hubungi tim admin melalui email support@efisonlt.com untuk permintaan instalasi versi lain.
Info Menjalankan GROMACS dengan Akselerasi GPU
ALELEON Supercomputer menyediakan GROMACS yang dapat berjalan dengan akselerasi GPU. Beberapa hal perlu menjadi pertimbangan user:
- Akselerasi GPU bukan jaminan peningkatan performa pada semua jenis input dan parameter.
- Tim admin menyarankan user untuk menguji hal ini di perangkat komputer pribadi.
- Biasanya GROMACS mdrun tidak memerlukan parameter tambahan untuk akselerasi GPU.
- Dalam beberapa kasus, optimasi parameter tambahan dapat meningkatkan kecepatan komputasi.
- Referensi lanjutan parameter akselerasi GPU (-nb, -bonded, -pme) dapat dipelajari pada tautan berikut:
Pre-processing Input dan Parameter
GROMACS membutuhkan pengolahan file input dan parameter sebelum menjalankan komputasi.
- Kegiatan ini dilakukan di login node sehingga tidak mengkonsumsi Kredit Core Hour.
- Hubungi tim admin apabila user membutuhkan panduan atau tambahan software package tertentu.
- Kegiatan ini dilakukan di terminal. Bagi user portal web EFIRO buka apps
Aleleon Shell Access.
ALELEON Supercomputer menyediakan variasi berikut:
gmx_mpi
GROMACS ALELEON Supercomputer diinstal dengan dukungan MPI sehingga user menggunakan gmx_mpi, bukan gmx.
| 1
|
Aktifkan modul GROMACS yang akan dipakai.
$ ml [nama-modul-GROMACS]
Nama modul lihat subbab 'Modul GROMACS' diatas.
|
| 2
|
Gunakan perintah gmx_mpi untuk pre-processing input.
Contoh dengan pdb2gmx:
$ gmx_mpi pdb2gmx [parameter-input-output]
|
ACPYPE (AnteChamber PYthon Parser InterfacE)
ACPYPE adalah wrapper Python berdasarkan software ANTECHAMBER untuk membantu pengolahan file input dan parameter software MD (Molecular Dynamic) seperti GROMACS ini. Tim admin menyediakan 2 opsi untuk menjalankan ACPYPE: (klik expand / kembangkan)
| Container ACPYPE siap pakai
|
| 1
|
Aktifkan container Apptainer:
$ ml apptainer
|
| 2
|
Aktifkan container ACPYPE, tersedia versi 2023.10.27:
$ apptainer run $SCONT/acpype-2023.10.27.sif
Shell masuk ke container tersebut dan user dapat menggunakan ACPYPE:
Apptainer> [jalankan perintah ACPYPE ...]
|
| 3
|
Untuk keluar dari container, jalankan perintah:
Apptainer> exit
|
| User Membuat Conda Environment* ACPYPE
|
| 1
|
Aktifkan Mamba:
$ ml Mamba/23.11.0-0
|
| 2
|
Buat conda env dengan environment ACPYPE yang disediakan:
$ mamba env create -f $CONDAYML/acpype.yml
- Perintah ini membuat conda env bernama acpype di HOME user.
- Conda env ini membutuhkan kapasitas storage 901MB.
|
| 3
|
Aktifkan conda env acpype:
$ source activate acpype
Shell masuk ke conda env tersebut dan user dapat menggunakan ACPYPE:
(acpype) $ [jalankan perintah ACPYPE ...]
|
| 4
|
Untuk keluar dari conda env, jalankan perintah:
(acpype) $ conda deactivate
|
| 5
|
| Untuk mengaktifkan kembali conda env lakukan langkah 1 dan 3.
|
* Note:
- Conda environment adalah ruang isolasi untuk instalasi library Python. Conda env ada karena karakteristik umum setiap komputasi Python (biasanya) membutuhkan instalasi library spesifik sehingga membutuhkan ruang isolasi untuk menghindari konflik library. Analogikan seperti anak sekolah mempunyai buku catatan tersendiri (conda environment) untuk setiap pelajaran (komputasi Python).
- Opsi conda environment mempunyai kelebihan yaitu user dapat mengendalikan instalasi versi ACPYPE dan depedencies-nya apabila dibutuhkan.
Pilihan Menjalankan Komputasi
User dapat menjalankan GROMACS dengan pilihan metode:
Batch Job
Menjalankan komputasi di compute node melalui manajemen Slurm kemudian user menunggu hingga selesai. Terdapat dua opsi tampilan yang dapat dipilih sesuai preferensi user: (klik expand atau kembangkan)
| Menggunakan Terminal
|
| 1
|
| Login SSH ke ALELEON Supercomputer.
|
| 2
|
| Siapkan file komputasi dan pre-processing input yang dibutuhkan.
|
| 3A
|
Buat Submit Script yaitu 'formulir' untuk menjalankan job komputasi.
- Nama file bebas dengan format .sh, contoh
submit.sh
|
|
|
Contoh template Submit Script, ikuti petunjuk NOTES didalamnya.
| Template Submit Script GROMACS versi CPU
|
#!/bin/bash
# -----------------------------------------------------
# Template SLURM Submit Script
# GROMACS (versi CPU) | rev.201224
#
# NOTES:
# 1. Isi bagian yang ditandai 4 garing (////).
# 2. Template ini bersifat referensi.
# User dapat mengubah bagian yang perlu diubah.
# -----------------------------------------------------
# -----------------------------------------------------
# Alokasi komputasi SBATCH dan file input
# -----------------------------------------------------
# Alokasi jumlah core thread CPU
#SBATCH --ntasks=////
# Alokasi jumlah memori RAM (satuan GB)
#SBATCH --mem=////GB
# Alokasi limit waktu menjalankan job
# Format HH:MM:SS atau D-HH:MM:SS
#SBATCH --time=////
# Definisi file untuk menampung output terminal program
#SBATCH --output=result-%j.txt
# Definisi file untuk menampung output error log
#SBATCH --error=error-%j.txt
# Parameter input dan output untuk gmx_mpi mdrun
GROMACS_PARAMETER="////"
# ----------------------------------------------------
# Script jalannya program
# ----------------------------------------------------
# Memuat modul software GROMACS
# Daftar modul lihat subbab "Modul GROMACS" di atas
module load gromacs////
# Perintah menjakankan GROMACS
mpirun -np ${SLURM_NTASKS} gmx_mpi mdrun ${GROMACS_PARAMETER}
|
| Template Submit Script GROMACS versi GPU
|
Note:
- GROMACS versi GPU menggunakan threading OMP per task MPI
- Task MPI diwakili oleh jumlah GPU (SBATCH gpus)
- Thread OMP diwakili oleh SBATCH cpus-per-task.
- Jumlah thread OMP disarankan 2 atau 4.
- Perhatikan total CPU = ntasks * cpus-per-task
|
#!/bin/bash
# --------------------------------------------------------
# Template SLURM Submit Script
# GROMACS (versi GPU) | rev.201224
#
# NOTES:
# 1. Isi bagian yang ditandai 4 garing (////).
# 2. Template ini bersifat referensi.
# User dapat mengubah bagian yang perlu diubah.
# 3. GROMACS GPU menggunakan threading OMP per task MPI
# Perhatikan usage CPU = ntasks * cpus-per-task
# --------------------------------------------------------
# -----------------------------------------------------
# Alokasi komputasi SBATCH dan file input
# -----------------------------------------------------
# Menggunakan partisi compute node GPU
#SBATCH --partition=ampere
# Alokasi jumlah core thread CPU
#SBATCH --ntasks=////
# Alokasi jumlah GPU, menjadi jumlah task MPI
#SBATCH --gpus=////
# Alokasi jumlah thread OMP per task MPI
# Disarankan 2 atau 4
#SBATCH --cpus-per-task=////
# Alokasi jumlah memori RAM (satuan GB)
#SBATCH --mem=////GB
# Alokasi limit waktu menjalankan job
# Format HH:MM:SS atau D-HH:MM:SS
#SBATCH --time=////
# Definisi file untuk menampung output terminal program
#SBATCH --output=result-%j.txt
# Definisi file untuk menampung output error log
#SBATCH --error=error-%j.txt
# Parameter input dan output untuk gmx_mpi mdrun
GROMACS_PARAMETER="////"
# ----------------------------------------------------
# Script jalannya program
# ----------------------------------------------------
# Memuat modul software GROMACS yang dapat menggunakan GPU
# Daftar modul lihat subbab "Modul GROMACS" di atas
module load gromacs////
# Threading OMP
export OMP_NUM_THREADS=${SLURM_CPUS_PER_TASK}
# Perintah menjalankan GROMACS 1 GPU dan multiple GPU
if [ $SLURM_GPUS -eq 1 ]
then
echo "Terdeteksi running GROMACS dengan 1 GPU"
gmx_mpi mdrun ${GROMACS_PARAMETER}
elif [ $SLURM_GPUS -gt 1 ]
then
echo "Terdeteksi running GROMACS dengan $SLURM_GPUS GPU"
# Unset UCX supaya MPI berjalan di NVIDIA
unset UCX_TLS
unset UCX_NET_DEVICES
mpirun -np $SLURM_GPUS gmx_mpi mdrun ${GROMACS_PARAMETER}
fi
|
|
- Untuk SBATCH notifikasi email status jalannya job lihat Slurm Jojo.
|
| 3B
|
SBATCH komputasi (ntasks, mem, time, dll) mempunyai limit berdasarkan:
- Sisa Core Hour user dan Fair Usage Limit.
- Spesifikasi sistem dan software komputasi.
Untuk melihat limit, jalankan perintah berikut:
$ slimit-gromacs
|
| 4
|
Jalankan job komputasi dengan perintah:
$ sbatch [nama-submit-script]
Contoh:
$ sbatch submit.sh
|
| 6
|
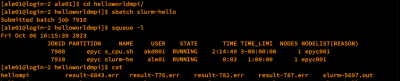
| User dapat melihat status jalannya job dengan perintah:
$ squeue -ul $USER
| Kolom ST atau STATE menunjukkan status jalannya job.
|
| STATE
|
Penjelasan
|
| R (RUN)
|
Job berjalan
|
| PD (PENDING)
|
Job tertahan, lihat NODELIST(REASON)
|
| CG (COMPLETING)
|
Job selesai dan dalam proses clean-up
|
| CA (CANCELED)
|
Job dibatalkan user
|
| PR (PREEMPETED)
|
Job dibatalkan admin, alasan dikabarkan via email
|
| S (SUSPENDED)
|
Job ditahan admin, alasan dikabarkan via email
|
|
|
|
Apabila user ingin menghentikan job yang berjalan, jalankan perintah:
$ scancel [job-ID]
Job ID ada pada squeue diatas.
contoh membatalkan job ID 231:
$ scancel 231
|
| Menggunakan Portal Web EFIRO
|
| 1
|
| Login ke web EFIRO ALELEON Supercomputer.
|
| 2
|
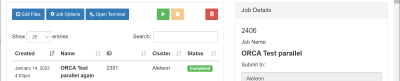
 Buka pinned apps Job Composer di homepage EFIRO. Buka pinned apps Job Composer di homepage EFIRO.
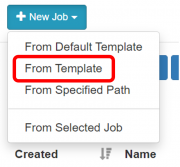
Pilih menu New Job -> From Template
|
| 3
|
| Pilih template GROMACS yang diinginkan:
|
| Nama Template
|
Penjelasan
|
| GROMACS 2021.1 (CPU)
|
Menjalankan GROMACS 2021.1 pada CPU
|
| GROMACS 2022.4 (CPU)
|
Menjalankan GROMACS 2022.4 pada CPU
|
| GROMACS 2022.4 (GPU)
|
Menjalankan GROMACS 2022.4 pada GPU
|
| GROMACS 2024.4 (CPU)
|
Menjalankan GROMACS 2022.4 pada CPU
|
| GROMACS 2024.4 (GPU)
|
Menjalankan GROMACS 2022.4 pada GPU
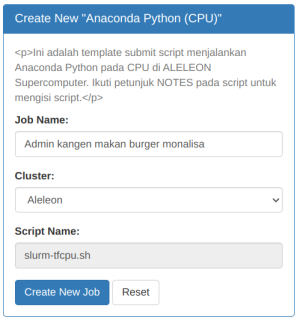
| Kemudian Isi Job Name dan klik Create New Job
|
| 4
|
| Siapkan file komputasi dan pre-processing input yang dibutuhkan.
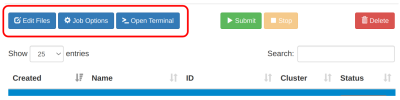
- Upload / download / edit file via menu
Edit Files
- Pre-processing input dan parameter via menu
Open Terminal.
|
| 5A
|
Lengkapi Submit Script melalui tombol Open Editor.
- Submit script adalah 'formulir' untuk menjalankan job komputasi.
- Ikuti petunjuk NOTES didalamnya.
- Klik
Save setiap kali mengubah script.
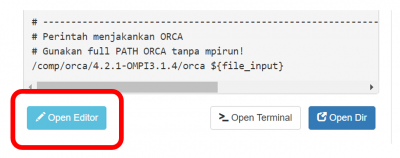
Note untuk GROMACS versi GPU:
- GROMACS versi GPU menggunakan threading OMP per task MPI
- Task MPI diwakili oleh jumlah GPU (SBATCH gpus)
- Thread OMP diwakili oleh SBATCH cpus-per-task.
- Jumlah thread OMP disarankan 2 atau 4.
- Perhatikan total CPU = ntasks * cpus-per-task
|
|
|
|
| 5B
|
SBATCH komputasi (ntasks, mem, time, dll) mempunyai limit berdasarkan:
- Sisa Core Hour user dan Fair Usage Limit.
- Spesifikasi sistem dan software komputasi.
Untuk melihat limit, jalankan perintah berikut melalui Open Terminal:
$ slimit-gromacs
|
| 6
|
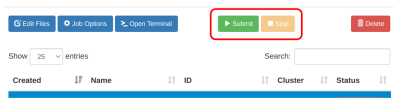
Jalankan job dengan klik tombol Submit.
- Pantau kolom status yang menjelaskan status jalannya job.
- Apabila ingin membatalkan job yang berjalan, klik
Stop.
| Daftar Status Job
|
| Status
|
Arti
|
| Not Submitted
|
Job belum pernah dijalankan.
|
| Running
|
Job berjalan.
|
| Queue
|
Job mengantri dan belum berjalan.
|
| Completed
|
Job selesai berjalan.
|
| Failed
|
Job berhenti di tengah jalan, antara error atau di stop user
|
Untuk melihat alasan job yang queue: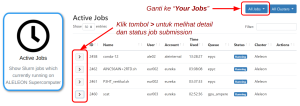 Buka pinned apps Active Jobs di homepage EFIRO. Ubah opsi All jobs ke Your Jobs. Klik simbol > untuk melihat status job yang pending pada kolom Reason. Arti reason lihat laman Daftar Reason NODELIST ALELEON Supercomputer. |
|
| 7
|
Lihat output file komputasi pada kolom Folder Contents
- Atau dengan membuka menu
Edit Files
|
| 8
|
Untuk menghapus ruang job dan datanya:
- Pilih ruang job yang akan dihapus.
- Klik menu
Delete
|
Pelaporan Kendala dan Support
Apabila menjumpai masalah teknis dalam menjalankan komputasi, silahkan lapor dengan klik gambar berikut ini.

---
Bila terdapat pertanyaan lainnya, silahkan hubungi admin EFISON melalui email:
support@efisonlt.com





 Buka pinned apps Job Composer di homepage EFIRO.
Buka pinned apps Job Composer di homepage EFIRO.








